



The perfect fold does not exist
Ramya Vishwanath explains the basics of protein folding, discussing the academic and clinical significance of the field, and where it is heading next
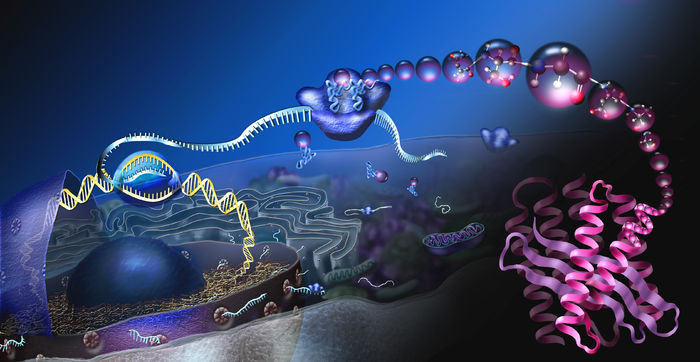
The human body consists of between 20,000 to over 100,000 unique proteins, and each one is folded perfectly. While most of us conjure images of food when we hear of proteins, as a biochemist, my brain has been re-wired to elicit a picture of a squiggly mass of strands condensed in a globular form. And in the truest sense, this is what proteins are – macromolecules consisting of long chains of amino acids folded in a distinct manner. Every protein has variable amounts of amino acids, usually fluctuating between fifty to two hundred. The large number of amino acids present gives rise to a plenitude of folding pathways of approximately ten to the power of three hundred. Yet, each time, the protein folds to only a single specific conformation. The folding process is spontaneous completed in a matter of milliseconds, and the precision and efficiency of this complex phenomenon are of great interest to scientists.
Protein folding is not only of academic interest. Comprehending the intricacies of this process holds immense value to understanding and finding the cure for several diseases, including cancer, type II diabetes, and Alzheimer’s. To understand the close link between protein folding – rather, protein misfolding – and disease, it is critical to evaluate the course and implications of it. The entire folding process occurs in steps, allowing the protein to evolve from the basic amino acid sequence called the primary structure to the folded state called the tertiary structure.
“Understanding how proteins fold could be the key to a throng of biological problems”
In the initial stage, the long straight chains of amino acids interact, leading to the formation of coils called alpha helices or zig-zag sheet-like structures called beta-sheets. In the second step, these alpha helices and beta-sheets interact to form larger and more complex structures having intricate, distinct folds in various regions. For instance, several beta-sheets may interlink to create a barrel-like structure or repeating units of interacting beta-sheets, and alpha helices may lead to the formation of a protein motif called the alpha/beta horseshoe. Thus, every protein has typical folding characteristics that define its structure and dictates its function. The structure-function correlation in proteins is well established. For instance, the correct folding of an enzyme allows it to condense into a structure where the active site is easily accessible. In the event of improper folding, the active site may not be generated or be difficult to access, thereby destroying the enzyme’s functionality. This dependence of the biological functionality of proteins on the final folded structure imparts paramount importance to the folding process, and understanding how proteins fold could be the key to a throng of biological problems.
The protein folding problem has resembled scattered pieces of a jigsaw puzzle for the longest time until research pieced together a few parts of the picture by running in vitro experiments. In 1962, Max Perutz and John Kendrew laid the foundation of the protein folding journey. While they successfully discovered the structure of haemoglobin using X-ray crystallography, they were unable to comprehend how the final folded structure was related to the simple amino acid sequence. In vitro experiments relied on techniques like circular dichroism and X-ray diffraction to establish protein structures. While these techniques had a high degree of accuracy, they were severely time-consuming. What followed was several years of arduous work, and while scientists were soon able to identify changes in protein structures and elucidate the final structure, they remained unsuccessful in monitoring the folding process.
In 1977, the first protein was studied via a computer simulation by a team at Harvard University. Protein folding researchers had begun to shift their focus from in vitro to in silico studies. Computer simulations allowed scientists to study the behaviour of macromolecules like proteins at a molecular level, and many believed computational techniques to be the final destination in the journey of the protein folding problem. However, understanding the folding mechanisms would involve simulating the entire macromolecule – a process that would entail writing the wave equations of every single atom in the protein. Thus, while running computer simulations was a reasonably fast way to study folding behaviour, it proved to be computationally intensive and expensive. However, the greatest disadvantage of computational methods was that they were considerably less accurate than lab-based methods. It was soon evident that relying on pure molecular simulation methods would not be a sustainable option in the long run.
“The use of AI and machine learning will fast-track research in this area”

Real breakthroughs have been possible only in the recent history by employing artificial intelligence assisted approaches. AI-based techniques are quick and accurate – they seem to combine the strengths of both lab-based methods and computational simulations. The most recent development in the field of protein folding is the invention of an AI programme, AlphaFold, by DeepMinds. When presented with the primary amino acid sequence structure of a protein, AlphaFold has the capacity to generate the folded structure with an accuracy that is almost comparable to painstaking lab-based methods. This discovery has significant implications. It has the potential to accelerate drug discovery and understand unfolding and misfolding mechanisms involved in diseases and find cures to them. The obvious next step is to harbour AI-based tools to investigate how proteins interact and combine to form larger entities.
The path of solving the protein folding problem has been complicated and challenging. While we still have a long way to go, the use of AI and machine learning will fast-track research in this area. It could very well transform protein folding sciences as we know it.
 News / C4P vandalises University offices over divestment10 June 2026
News / C4P vandalises University offices over divestment10 June 2026 Lifestyle / A town with a view11 June 2026
Lifestyle / A town with a view11 June 2026 Comment / Top of the slops: the competitiveness of college dining4 June 2026
Comment / Top of the slops: the competitiveness of college dining4 June 2026 Sport / The imperfect privilege of Cambridge sport12 June 2026
Sport / The imperfect privilege of Cambridge sport12 June 2026 Comment / ‘On the Poverty of Student Life’: sixty years on10 June 2026
Comment / ‘On the Poverty of Student Life’: sixty years on10 June 2026
 Music / Hejira: a jazzed tribute to Joni Mitchell 13 June 2026
Music / Hejira: a jazzed tribute to Joni Mitchell 13 June 2026 Film & TV / Lights, camera, Cambridge13 June 2026
Film & TV / Lights, camera, Cambridge13 June 2026 Lifestyle / Confessions of a May Ball crasher12 June 2026
Lifestyle / Confessions of a May Ball crasher12 June 2026 Lifestyle / May Week horoscopes12 June 2026
Lifestyle / May Week horoscopes12 June 2026 Lifestyle / The finalist’s bucket list12 June 2026
Lifestyle / The finalist’s bucket list12 June 2026



